The Howlett Lab is located in the Department of Cell and Molecular Biology at the University of Rhode Island. We study the eukaryotic cellular DNA damage response and the maintenance of genome stability. We study these topics primarily through the lens of a rare genetic disease, Fanconi anemia.
Fanconi anemia (FA) is clinically characterized by congenital malformations, increased risk for bone marrow failure and cancer, and premature mortality. Several recent clinical reports have described pleiotropic neurological symptoms in FA patients, including abnormal brain MRIs, seizures, brain lesions, and early-onset cognitive decline. This constellation of neurological symptoms is collectively referred to as Fanconi Anemia Neurological Syndrome (FANS). The molecular etiology of FANS is unknown.
FA is both autosomal and X-linked (FA complementation group B is caused by mutation in the X-linked FANCB gene). The incidence of FA in the U.S. is approximately 1 in 120,000 live births. FA is caused by mutation of any one of 23 genes: FANCA, FANCB, FANCC, BRCA2/FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, BRIP1/FANCJ, FANCL, FANCM, PALB2/FANCN, RAD51C/FANCO, SLX4/FANCP, ERCC4/FANCQ, RAD51/FANCR, BRCA1/FANCS, UBE2T/FANCT, XRCC2/FANCU, MAD2L2/FANCV, RFWD3/FANCW, and REV7/FANCY.
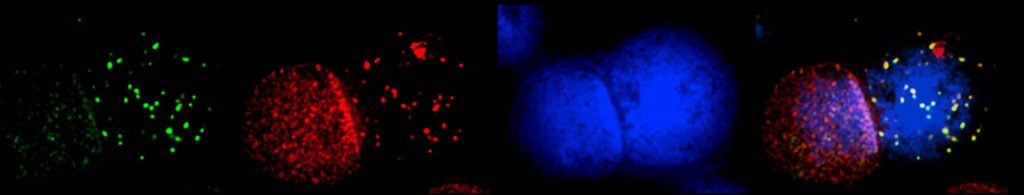
In vitro studies have established that the FA proteins function together to orchestrate the repair of DNA interstrand crosslinks (ICLs), induced by DNA crosslinking agents such as mitomycin C and cisplatin. However, the endogenous source of genome instability for FA patients remains to be elucidated. Recent studies have strongly suggested that reactive aldehydes generated through normal metabolic processes, e.g., acetaldehyde and formaldehyde, may be particularly genotoxic to FA patient cells.